钴酸锂
钴酸锂
点击蓝字 关注我们
钴酸锂是第一款商业化锂离子电池的正极材料,其完全脱锂后的理论克容量为274mAh/g,真密度高达5.1g/cm3,实际压实密度可达4.2g/cm3,具有极高的体积能量密度(高电压下优势凸显),目前仍然是消费类电池应用最广泛的正极材料。
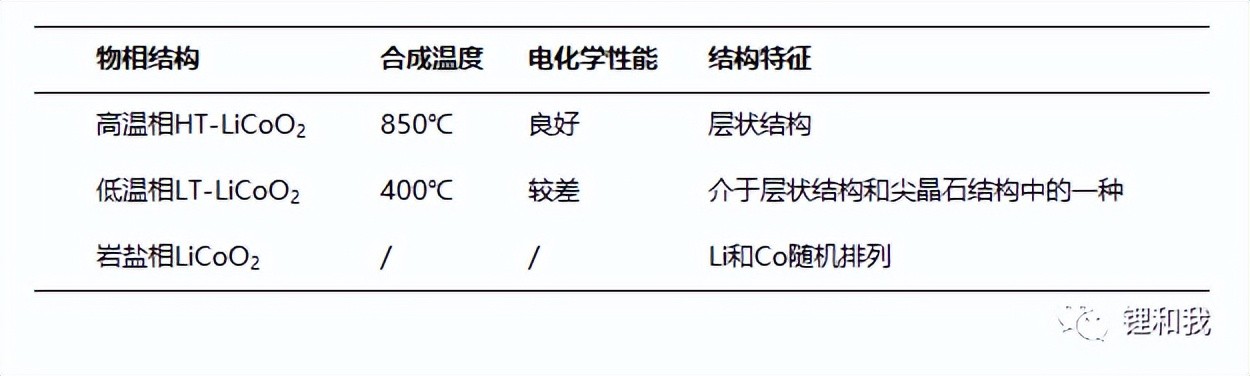
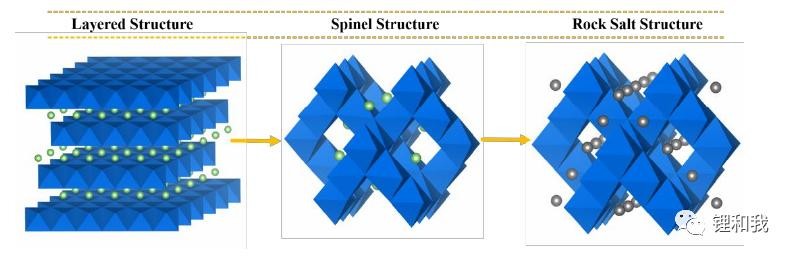
实际上,钴酸锂具有三种晶体结构,分别为高温相HT-LiCoO2、低温相LT-LiCoO2、岩盐相LiCoO2。其中,低温相钴酸锂合成温度较低,晶体结构特征介于层状结构和尖晶石结构之间,Li层中含有约25%Co原子,Co层中含有约25%Li原子,松装密度较低,电化学性能较差,很少作为商业化正极材料投入使用,而岩盐相钴酸锂的结构高度紊乱,Li和Co在晶体内部随机排列,没有明显规律。
钴酸锂的三种晶体结构简介
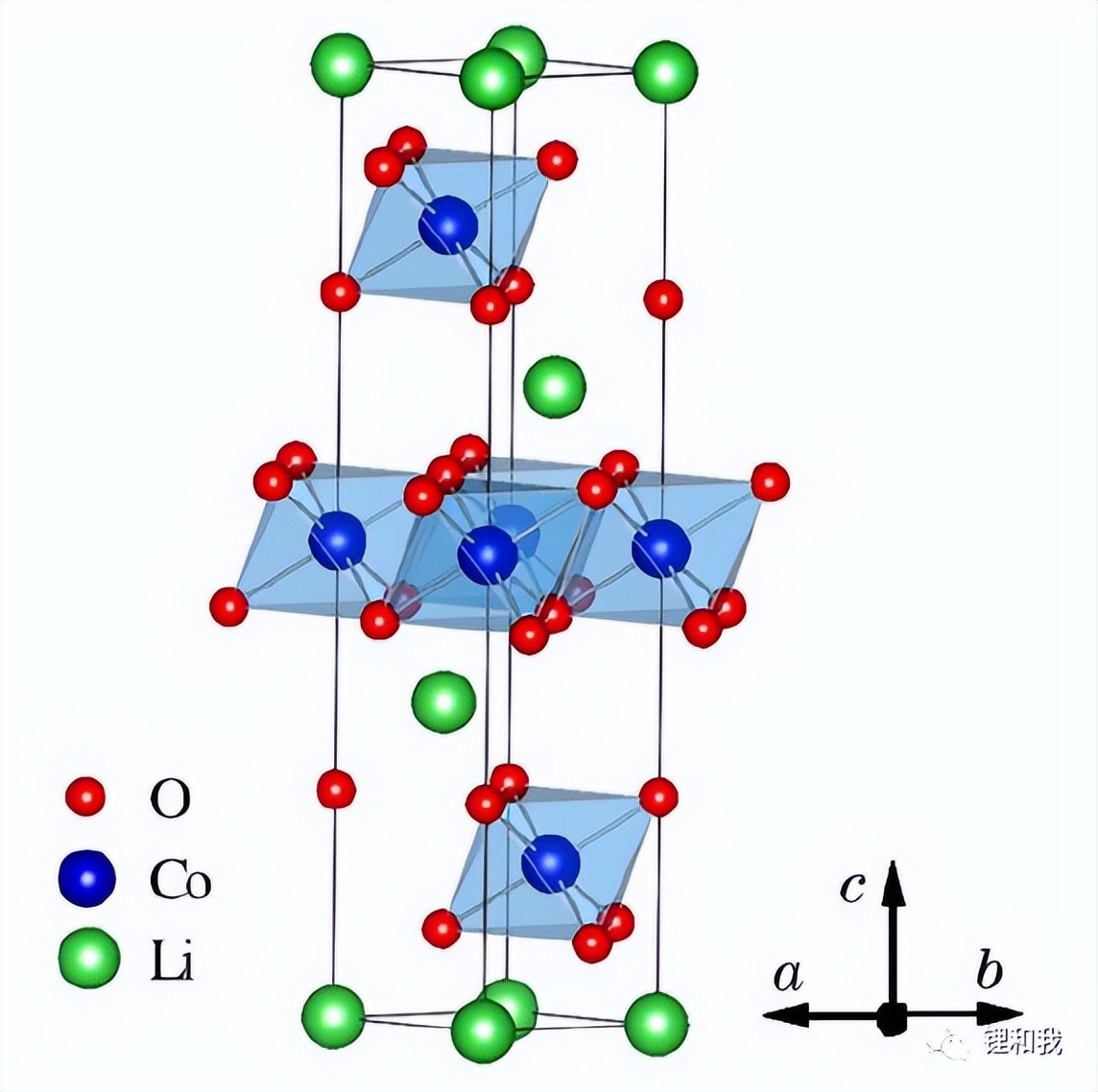
目前通常所说的钴酸锂就是指高温相HT-LiCoO2,其隶属于α-NaFeO2层状结构六方晶系,空间群为R-3m,Co原子与相邻的O原子通过共价键形成CoO6八面体,Li原子与相邻的O原子通过离子键形成LiO6八面体,Li+和Co3+交替排列在O2-形成的骨架结构中,形成“-O-Li-O-Co-O-Li-O-Co-”排列结构,由于Co-O键作用力强于Li-O键,因此,有助于充放电过程中Li+在CoO2层间脱出和嵌入,钴酸锂的层状结构不易坍塌,从而保证了材料具有较好的循环稳定性。
钴酸锂的晶体结构模型
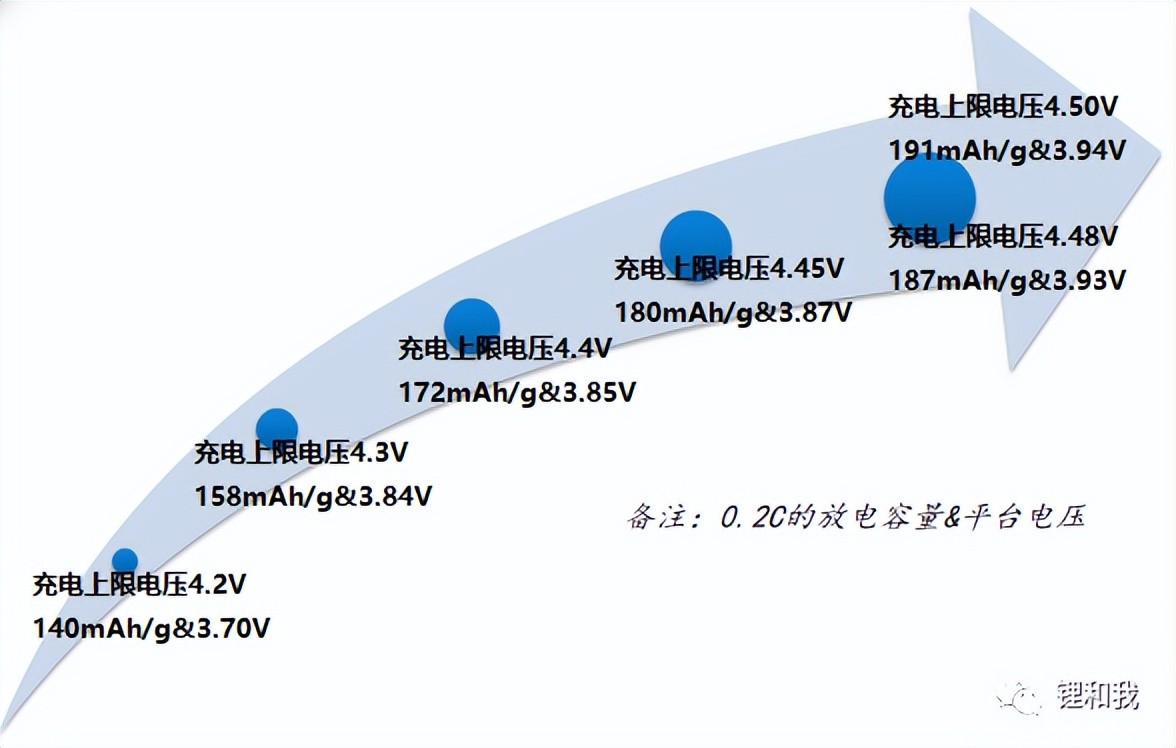
更高的能量密度是锂离子电池不懈的追求。提高充电上限电压可以使钴酸锂脱出更多的Li+参与电化学反应,从而提升全电池的放电比容量和放电平台,如将钴酸锂充电上限电压从4.2V提高到4.45V,放电比容量从140mAh/g提升到180mAh/g(提升约28.6%),放电平台从3.70V提升到3.87V(提升约4.6%),因此,提高钴酸锂充电上限电压是提升电池能量密度最有效的方法之一。
钴酸锂不同充电上限电压对应的比容量和放电平台
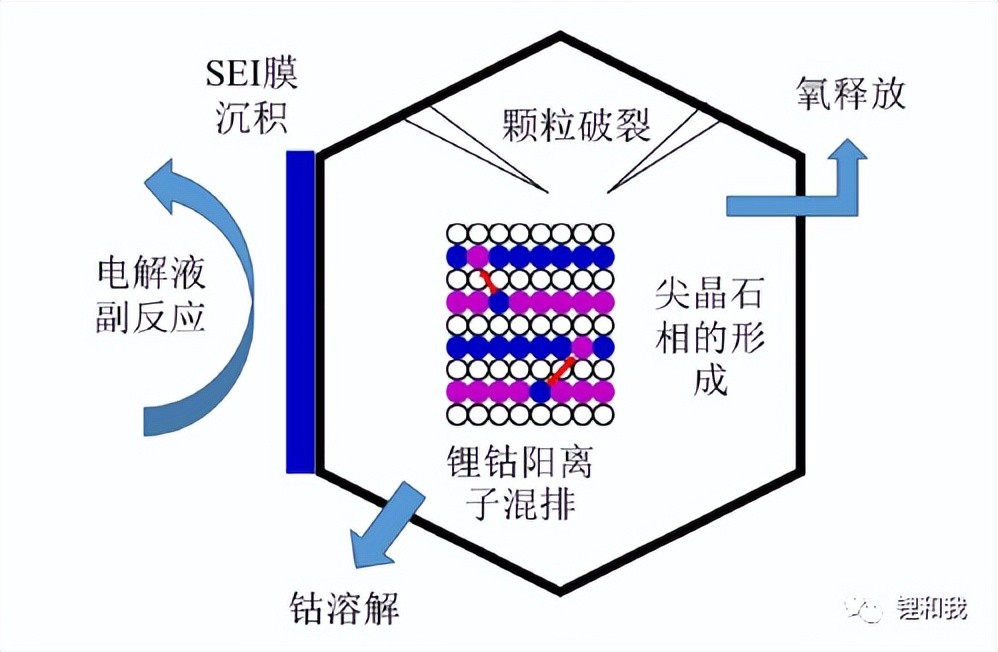
然而,提升充电上限电压后(过量脱锂)会带来一系列问题,如材料相变、界面副反应、钴金属溶出、氧气析出等,导致材料性能尤其是循环性能快速衰减。钴酸锂表面反应活性高于体相,在其表面的充电过程包括如下反应步骤:
step1:钴酸锂表面位置优先脱出Li+;
step2:Li+脱出后O原子间失去阳离子阻隔产生排斥,表面结构开始变得不稳定;
step3:Li+持续脱出,表面处晶格氧活性提高到一定程度发生析氧;
step4:析氧发生后,表面的Co原子稳定性变差,发生溶解;
step5:高价元素Co4+同时氧化电解液,直接参与化学反应溶入电解液。
钴酸锂正极容量衰减机制
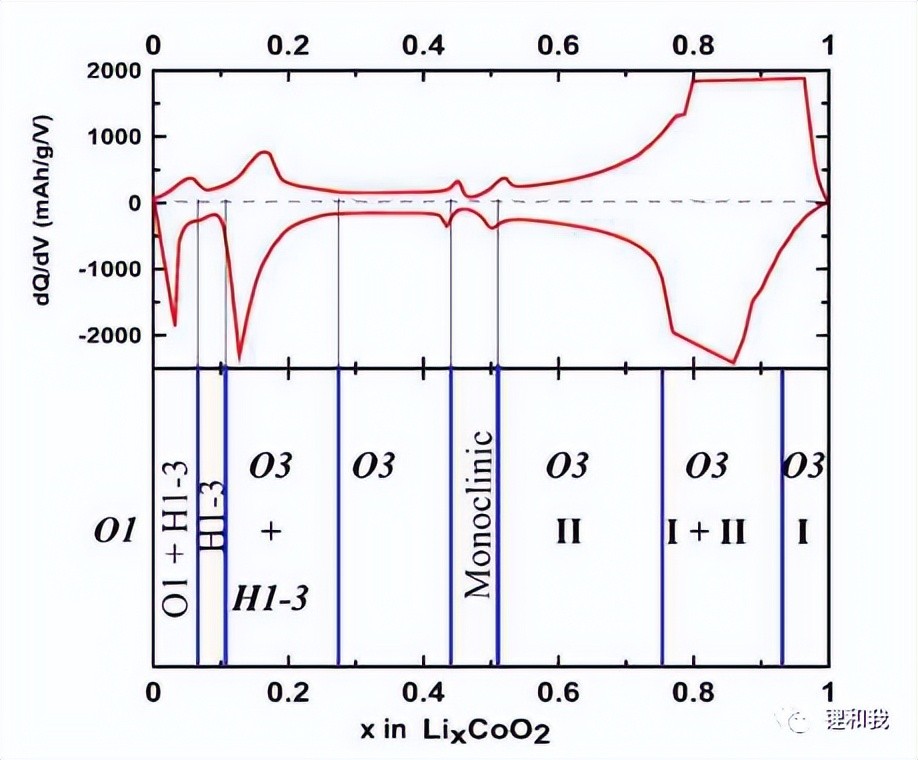
如下图所示,在充电过程中,随着Li+从钴酸锂中脱出,材料的晶体结构发生改变,导致材料结构发生不可逆破坏,Li+难以进行可逆脱嵌从而造成容量损失。在钴酸锂充电过程中,存在两次向单斜晶系的相变过程,当充电到4.2V时,Li+脱出量约50%,此时,钴酸锂由六方晶系(H-2相)转变为单斜晶系(M相),Li+脱出后相邻O原子层由于静电排斥力比Li-O静电吸引力更大而使材料晶胞c轴膨胀约2.3%,当钴酸锂充电到4.5V以上时,钴酸锂由六方晶系(O3相)转变为单斜晶系(O1相),材料晶胞c轴膨胀约2.6%,相比于4.2V时提高了13%,巨大的晶胞体积变化容易导致材料层状结构坍塌,产生颗粒裂纹甚至粉化,部分Li+无法回嵌到CoO2层,最终导致循环容量保持率快速衰减。
LixCoO2(0≤x≤1)的微分容量随Li+浓度变化关系
钴酸锂上限电压提高后带来的第二个问题是界面副反应增多。高电压下电解液的电化学稳定性变差,容易分解产生HF,从而对SEI膜造成腐蚀,进而影响Li+的脱嵌。其次,高脱锂态下的钴酸锂含有强氧化性的Co4+,电解液在钴酸锂表面被氧化产生副产物,从而增大界面阻抗,最终导致容量加速衰减。
反应机理如下:
LiPF6→LiF(s)+PF5
H2O(微量水)+PF5→POF3+2HF
电解液锂盐LiPF6是一种极不稳定的物质,在高电压下加速分解产生不溶于电解液的LiF和高反应活性的路易斯酸PF5,然后与电解液中的微量水反应生成HF。研究表明,端羟基LiCoO2对HF具有高反应活性,从而生成H2O和LiF沉淀,附着在电极表面,阻碍Li+在正极材料和电解液界面之间迁移,导致电池容量衰减。
第三个问题是钴金属溶出和析氧问题。随着Li+的持续脱出,钴酸锂表面Co元素和O元素活性进一步增强,当晶格氧活性提高到一定程度时,就会以O2的形式逸出,气体逸出后,Co原子稳定性变差,这一过程会伴随Co溶解。
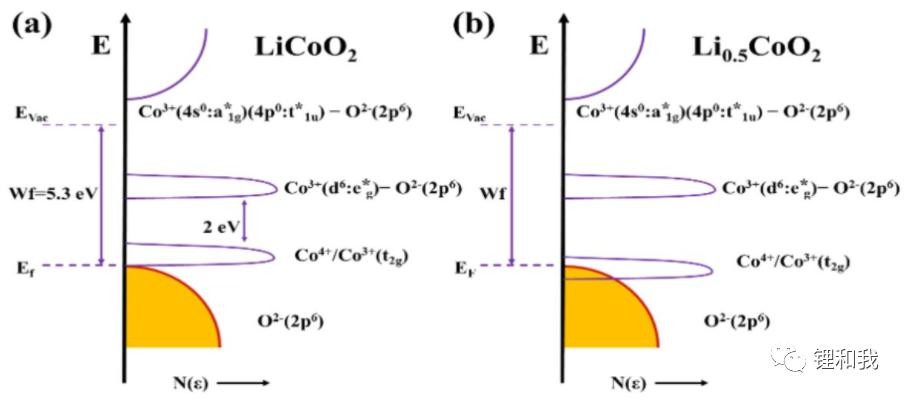
在常见的过渡金属元素中,相比于Mn、Ni等元素,Co与O具有最强的相互作用,二者的电子轨道重合最为明显,随着充电上限电压提高,正极材料失去的电子逐渐增加,当Co元素高于O元素2p轨道的价带顶电子消耗殆尽时,进一步的电荷补偿将会发生在Co的t2g轨道和O的2p轨道重叠区域,即发生在这一区域的电荷补偿不再由Co元素独立提供电子,阴离子O元素也会参与其中。从下图钴酸锂的电子能带结构图可以看出,钴酸锂未脱锂时,Co4+/3+(t2g)能带和O2-(2p)能带是分离的,而当钴酸锂中有一半Li+脱出时,Co4+/3+(t2g)能带和O2-(2p)能带存在部分重叠,晶格O参与电荷补偿,进一步脱锂时,就会导致O2逸出和Co溶解,材料骨架结构稳定性变差,最终导致电化学性能和安全性能的衰减。
钴酸锂的电子能带结构图
钴酸锂因其自身材料结构原因,高电压下(过量脱锂)容易导致不可逆相变发生,层状结构不稳定,伴随着晶面滑移,原子重排导致晶胞参数剧变,晶界产生错位,表面应力变化造成颗粒破裂,电解液氧化分解产生副反应,氧气逸出,钴金属溶解等等问题导致电池性能失效。目前,针对材料本身,主要通过表面包覆和体相掺杂等方式对钴酸锂进行改性,从而改善其在高电压下的稳定性。
钴酸锂材料常见的改性思路
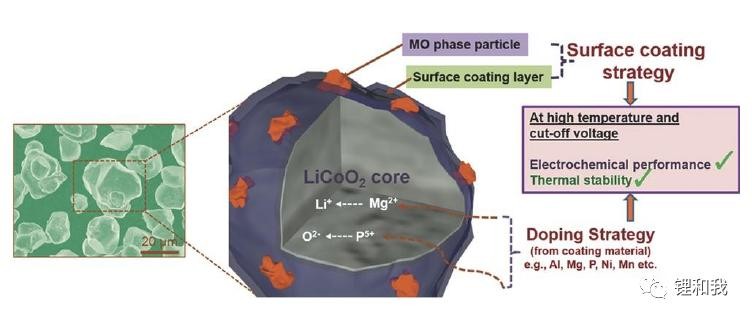
表面包覆主要是改变钴酸锂表面的物理及化学状态,在一定程度上稳定其材料形貌及晶体结构。通过表面包覆处理可以在钴酸锂材料表面构筑一层“屏障“,稳定活性材料与电解液的界面,防止HF对电极材料的腐蚀,抑制钴溶出和电解液氧化等界面副反应。体相掺杂主要是改善钴酸锂的结构稳定性,是对钴酸锂本征性质的改善。掺杂元素进入Li、Co、O位置后,由于掺杂元素的差异性,会改变其占据位点的原子状态,进而改变材料整体的原子排列和电子云分布,进而影响材料性能,是一种克服材料自身缺陷优化材料性能的重要方法。
针对钴酸锂在4.2V的钴溶解、电解液氧化分解和材料相变问题、可以通过表面包覆予以有效解决。而在4.5V以上的材料相变问题和氧流失问题,多元素协同掺杂是一种更有效的改性策略(如主族元素掺杂:Na、Mg、Ca、Al、Cu等,过渡金属元素掺杂:Ni、Mg、Ti、Zn、Cr、Fe等,非金属元素掺杂:B、P、Si、F等,稀土元素掺杂:Nd、La等),但体相掺杂对结构稳定性的改善并不是影响高电压钴酸锂性能的唯一因素,需要结合表面包覆来抑制界面副反应,钴溶解和氧气逸出。因此,采用单一方式可能无法兼顾材料结构和界面问题,将两种改性方式有效结合,是提升高电压钴酸锂性能的重要技术手段。
表面包覆技术主要包括四大类:
1)电子导体包覆;
2)离子导体包覆;
3)电子、离子双导体包覆;
4)电子、离子双绝缘包覆。
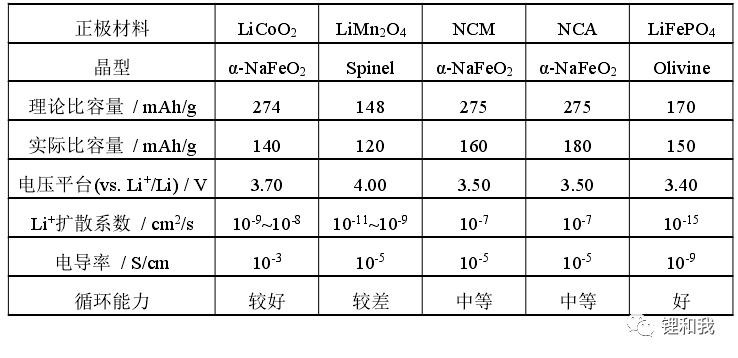
电子导体包覆主要包括C包覆及其他电子电导较高的化合物包覆,但由于钴酸锂本身具有半导体特性,且在合成过程中难免产生Li空穴而使其电子电导率十分优异,是目前锂离子电池正极材料中电子电导率最高的材料,因此并不需要做特别的改善,此外,高温下C会将钴酸锂还原为CoO和Co3O4,因此技术层面也难以应用。由于LFP具有较低的电子电导率和离子电导率,C包覆在LFP上的应用最为成功,可以显著提升其电化学性能。
常见正极材料性能对比
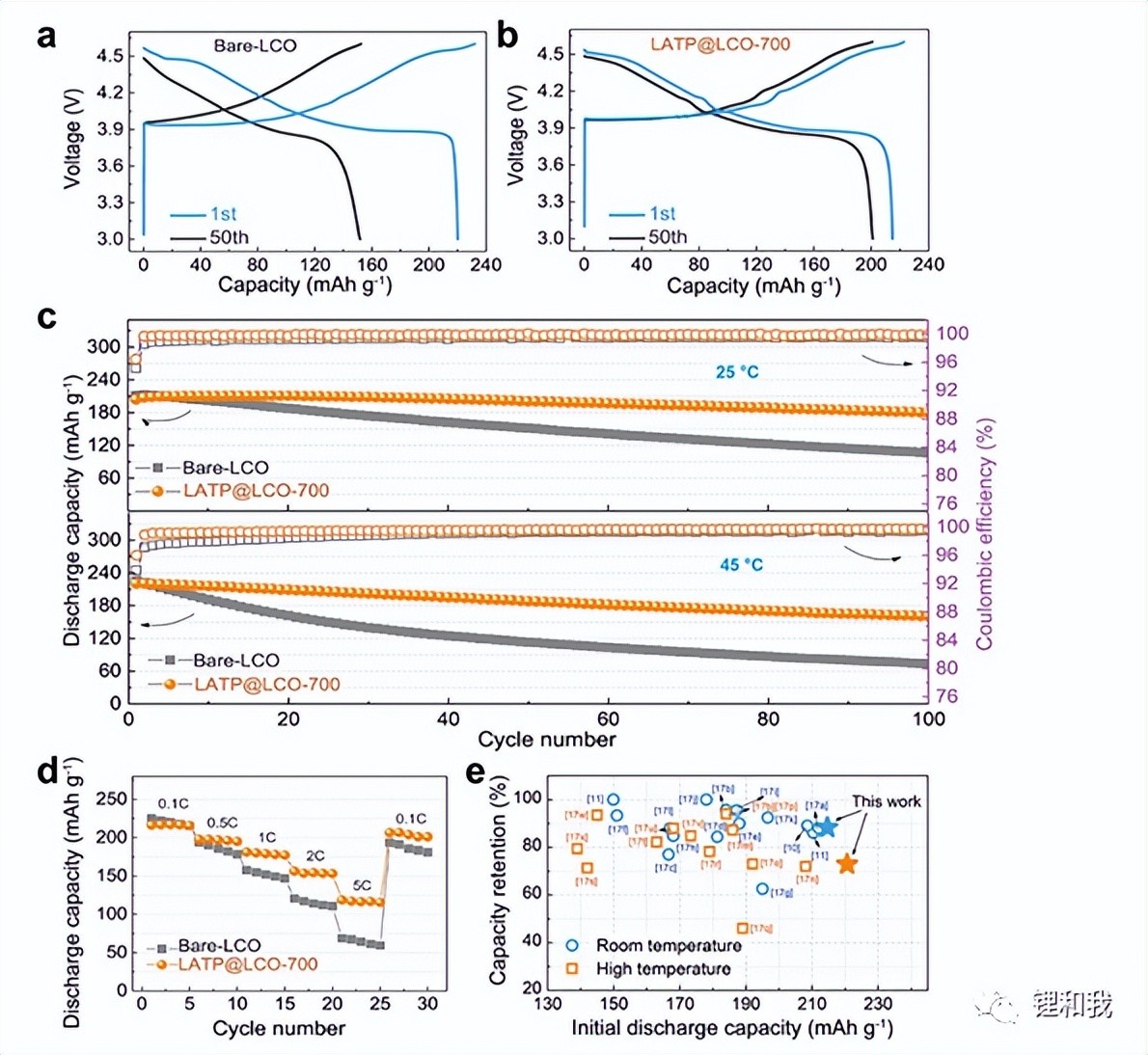
离子导体包覆是在钴酸锂表面包覆一层具有较高离子电导的材料,由于固体电解质材料通常具有较高的离子电导和较宽的电化学窗口,常用作离子导体包覆材料。如Li1.5Al0.5Ti1.5(PO4)3(简称LATP)包覆在钴酸锂表面,可以大大提高4.5V下的循环稳定性,同时对材料倍率性能也有一定改善。但是,电子绝缘、质量较重的固体电解质包覆也可能导致材料内阻增加、比容量降低等问题,因此,超薄连续的固体电解质包覆应该是主要的技术方向。
LATP包覆前后的钴酸锂性能对比
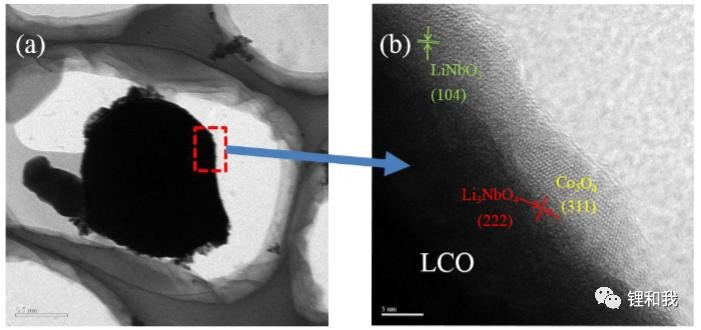
电子、离子双导体包覆是指包覆层既能传导电子,又能传导锂离子的材料,这类材料通常也可以独立用作电极活性材料,如LiMn2O4、LiFePO4、Li2TiO3、Li4Ti5O12、LiNbO3等。最早报道的电子、离子双导体包覆材料是锰酸锂,当时应用在4.3V钴酸锂材料上,取得了突破性的进展,随后,比容量更高的磷酸铁锂成为了更合适的选择,同时其优异的稳定性可以保护正极表面免受电解液腐蚀,在4.2V下,LFP包覆对循环稳定性和高温性能提升非常显著,而在更高电压下(如4.5V以上),LiNbO3包覆的钴酸锂表现出了更加优异的循环性能。
LiNbO3包覆的钴酸锂HRTEM图像(三相混合包覆层)
电子、离子双绝缘包覆看起来似乎除了具有物理阻挡外,不会对电池性能产生其他有益的影响,然而事实并非如此,目前最常见和最理想的包覆方式就是电子、离子双绝缘包覆,如氧化物(Al2O3、MgO)、氟化物、磷化物。
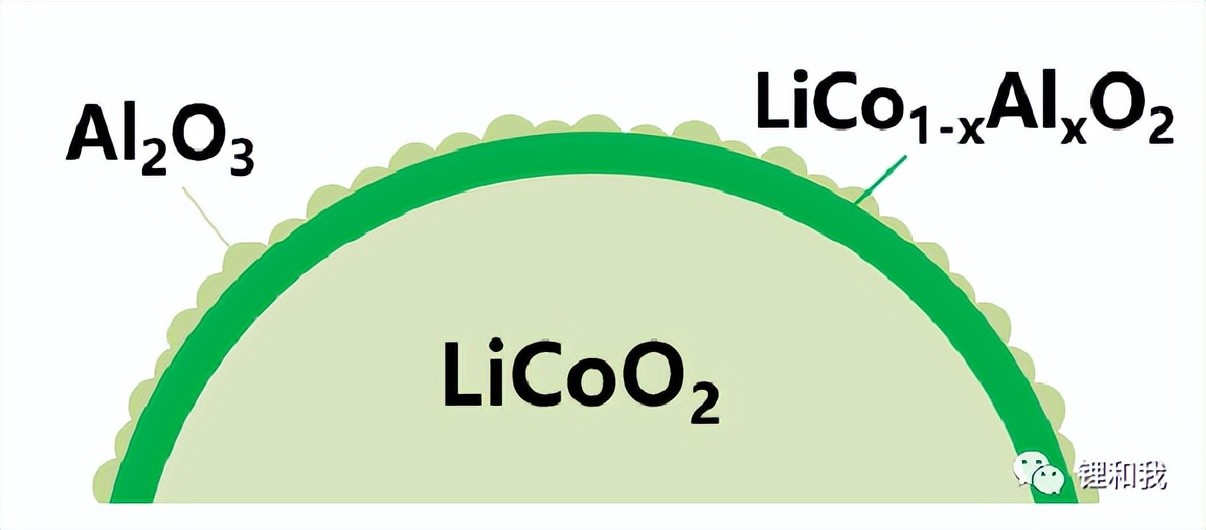
学术界关于氧化物包覆对材料性能改善的一致观点是:氧化物包覆能够实现物理阻挡与化学稳定,缓解Co溶解。同时,包覆的氧化物具有双性氧化特性,既能与酸反应,又能和碱反应,可以消耗电池体系中的HF等腐蚀性物质,提高活性材料界面稳定性。但目前对于氧化物包覆还存在两个争论点:一是包覆的连续性问题,与电子、离子双导体包覆不同之处在于,氧化物本身不导电,完全致密或厚度超过电子隧穿距离的包覆层都会造成活性材料电化学性能下降,,如何掌握包覆层的厚度和连续性是一大难点;二是包覆物是以何种形式存在,是以无机物的形式存在还是与钴酸锂形成固溶体,目前尚存争论。
Al2O3在钴酸锂表面可能的存在形式
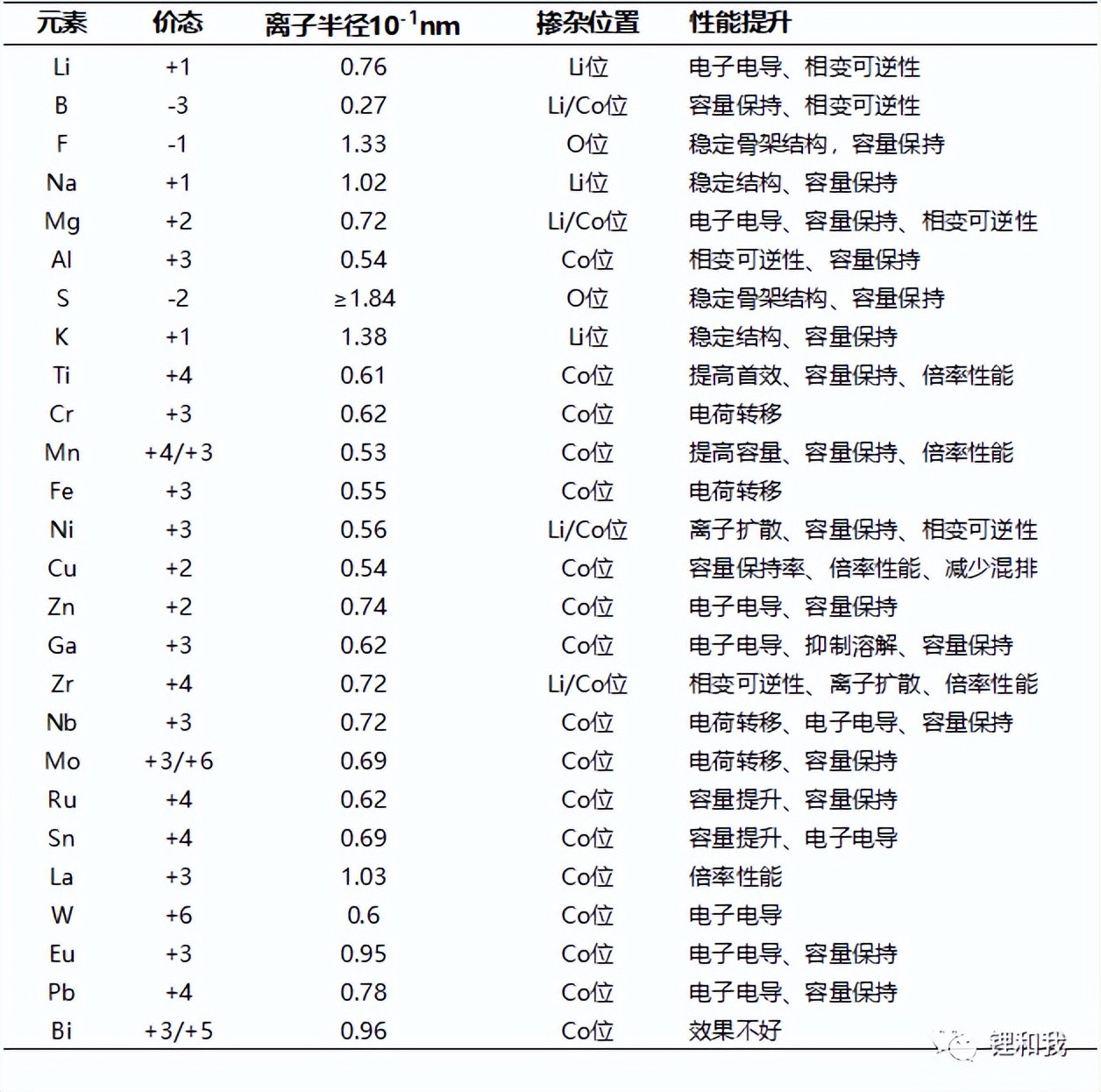
体相掺杂是一种常见且有效的改性方式,在合成钴酸锂的过程中,加入其他元素,能够使材料具有新的物理和化学特性,且掺杂工艺简单、可控性强,能大规模商业化制备。钴酸锂的掺杂改性主要通过三方面调控:
1)掺杂元素类型;
2)掺杂元素比例;
3)掺杂元素位置。
钴酸锂常见掺杂元素的性质及作用
在众多元素中,Mg、Al、Ti是商业化钴酸锂最常用的三种掺杂元素,但在实际应用中,为了确保最大化发挥正极材料的性质,掺杂元素含量通常较少,一般是几百至几千ppm,百分比含量通常不超过1%。为了系统研究这三种掺杂元素对钴酸锂性能的影响,分别设计了单元素掺杂、双元素掺杂和三元素共掺杂,具体做法是将合成钴酸锂的前驱体Li2CO3、Co3O4和掺杂元素的氧化物按照预设比例混合后加热烧结得到一次烧结物,然后将粉末研磨充分后再进行二次烧结,得到最终产物。
钴酸锂元素掺杂过程
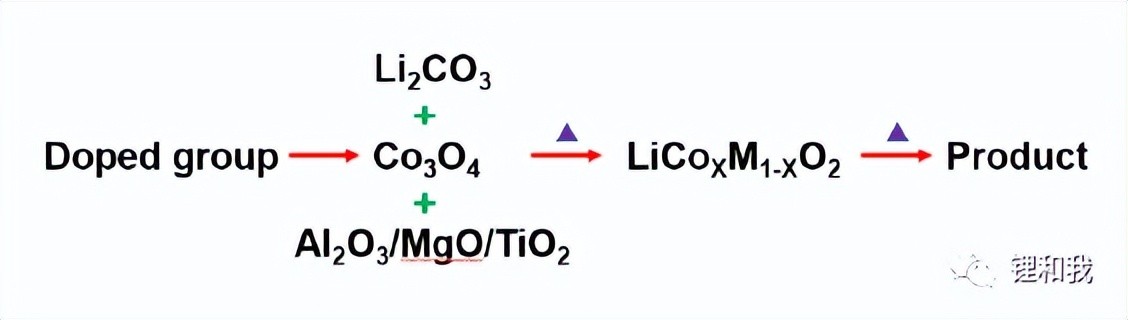
通过ICP对掺杂元素含量进行测定结果如下,证明了钴酸锂材料中含有掺杂的目标元素,并且不存在交叉污染现象。
钴酸锂元素掺杂质量百分比
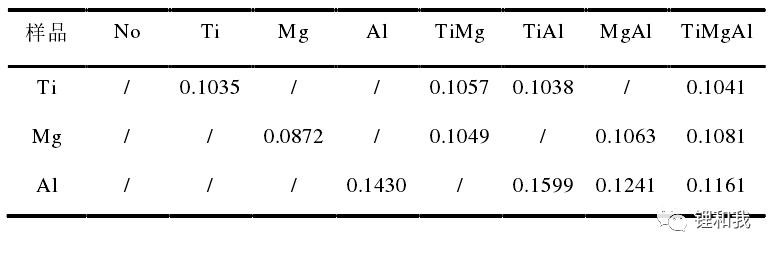
然后对其进行粒径测试,判断掺杂过程是否会对材料粒径产生影响,结果表明,在第一次烧结过程中加入掺杂元素对钴酸锂粒径分布存在明显影响,可能抑制了钴酸锂的晶体生长,但Mg、Al、Ti三种元素对比发现,Mg和Al元素对粒径影响较小,而Ti元素掺杂后钴酸锂粒径减小了近一倍,这一现象可能与Ti元素的离子半径有关(介于Li和Co离子半径之间),因此不易掺杂进入材料晶格中,从而引起了材料内部应力的变化。
钴酸锂元素掺杂后的粒径分布(单位:um)
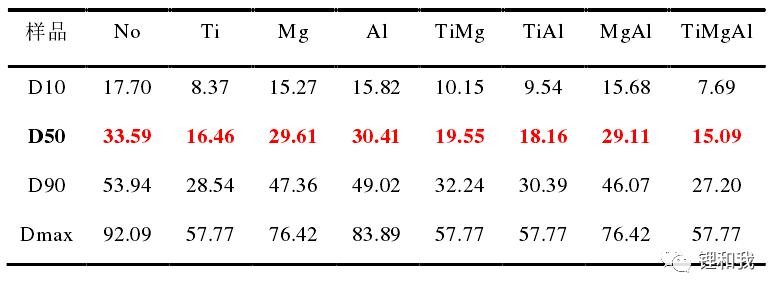
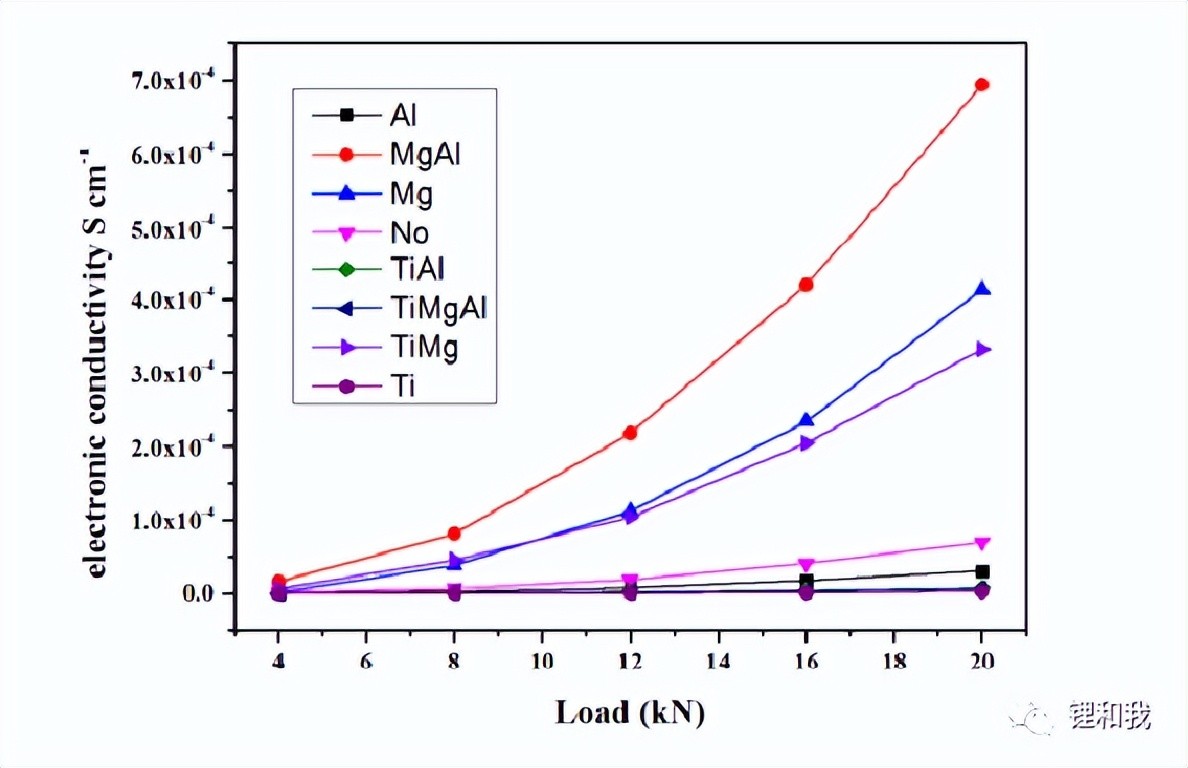
对制备的钴酸锂粉末进行压片后采用四探针法测试电导率,发现所有含Mg元素的钴酸锂,其电子电导率均比未掺杂钴酸锂更高,而其他元素掺杂的钴酸锂,其电子电导率均比未掺杂钴酸锂更低。这是因为低价的Mg2+掺杂破坏了Co3+周围的电荷守恒,产生了部分Co4+,从而提高了材料的电子电导率,而高价Ti4+掺杂则会降低这一效果。
钴酸锂不同元素掺杂后的电子电导率
通过电化学性能测试发现:Mg元素掺杂能够提高钴酸锂的电子电导并能协助Al元素更好的提高材料循环稳定性,Al元素对钴酸锂晶体内部的应力最小,有利于循环稳定性,而Ti元素掺杂可以显著提高钴酸锂的首次效率和放电比容量。
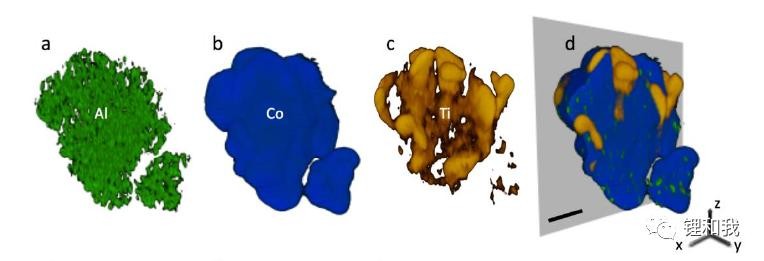
应用同步辐射X射线扫描透射成像技术对掺杂不同元素的钴酸锂进行分析,发现由于Co元素是构成材料的主体元素,均匀且致密的存在于颗粒之中,Al元素含量较少,均匀分布点缀在钴酸锂颗粒的各个部位(表面和内部),而Ti元素在钴酸锂颗粒表面出现了富集现象,表明该元素并未能完全掺杂进入钴酸锂晶格中。
钴酸锂不同元素掺杂的位置解析
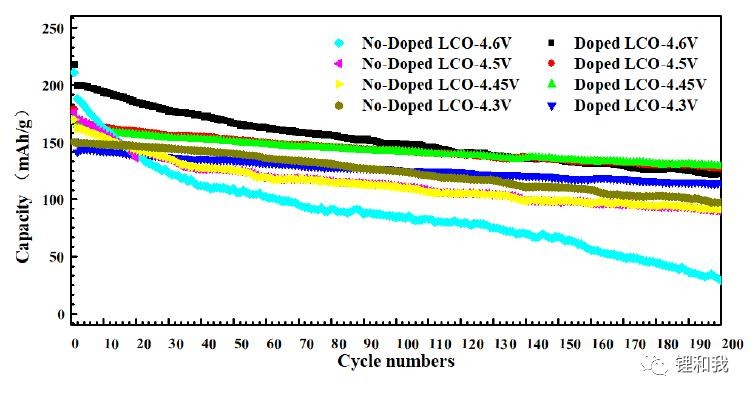
为了验证元素掺杂对高电压钴酸锂长期循环的影响,分别选择了4.3V、4.45V、4.5V和4.6V上限电压进行循环,可以发现,元素掺杂后的钴酸锂在不同上限电压下的循环性能均得到了提高,证明了元素掺杂对改善高电压钴酸锂的巨大潜力。尽管如此,其在4.6V下的循环性能仍然较差,循环200周后的剩余容量仅相当于4.3V钴酸锂的水平,由此可见,4.6V钴酸锂的商业化应用仍然任重道远。
未掺杂元素的钴酸锂和Mg、Al、Ti三元素共掺杂的钴酸锂在不同上限电压下的循环性能对比
钴酸锂在4.5V以上高电压下的容量衰减与氧的电荷补偿行为以及不可逆相变密切相关,且电压越高,氧的电荷补偿行为越严重,严重破坏了晶格氧的稳定性,还为高电压钴酸锂的安全性能埋下了隐患,同时,不可逆的相变过程也会导致容量迅速衰减。因此,如何抑制氧参与电荷补偿以及如何稳定材料晶体结构,是进一步优化高电压钴酸锂电化学的关键突破口。
更多内容请关注微信公众号 ☞
标签: